- Abbildung 1:
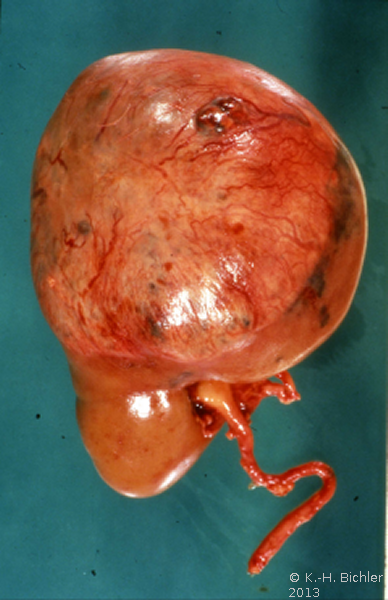
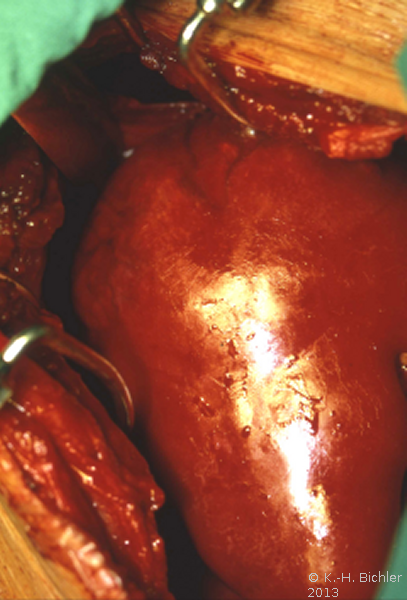
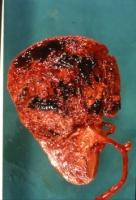
- a) Intraoperativer Situs: Ausgedehnter Tumor


Die häufigste morphologische Form des Nierenzellkarzinom beim Erwachsenen ist mit 70% das klarzellige Karzinom gefolgt vom papillären (chromophilen) und chromophoben Tumor mit 27%. Beim NCC im Kindes- und Jugendalter werden klarzellige und der papilläre Tumor gefunden. Estrada et al fanden unter 11 Nierenzellkarzinomen im Kindes- bzw. Jugendalter 5 papilläre und 6 klarzellige ![]() Literatur:Estrada C. R. et al: "Renal Cell Carcinoma: Children's Hospital Bosten Experience", Pediatric Urology, 66, 1296-1300, 2005. Darüber hinaus gibt es seltene Tumore wie das Collecting-Duct-Karzinom (Duct-Bellini-) bzw. Onkozytome (s. Kasuistik 2).
Literatur:Estrada C. R. et al: "Renal Cell Carcinoma: Children's Hospital Bosten Experience", Pediatric Urology, 66, 1296-1300, 2005. Darüber hinaus gibt es seltene Tumore wie das Collecting-Duct-Karzinom (Duct-Bellini-) bzw. Onkozytome (s. Kasuistik 2).
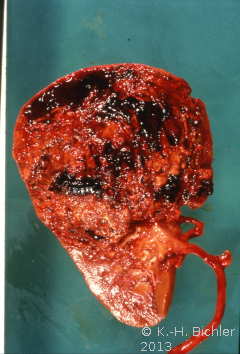
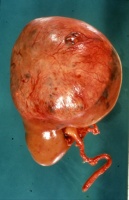
Makroskopisch zeigt der Tumor eine gelborange Farbe und ist durch Einblutungen und weißliche, Nekroseherde gesprenkelt. Außerdem finden sich Zysten von Millimeter- bis Centimeter-Größe (Abbildung 1). Auffällig häufig enthält das Nierentumorgewebe Kalzifizierungen. Zumeist ist der Tumor mehr oder weniger gekapselt und dadurch vom Nierenparenchym abgetrennt (s. Abbildungen Kasuistiken).
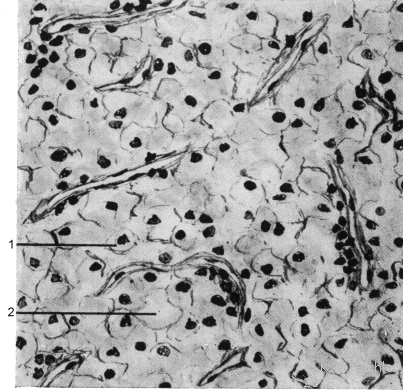
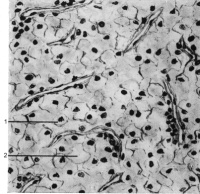
Mikroskopisch wird das Bild von klarzelligen, zum Teil drüßenschlauchähnlichen (tubulären) oder soliden Strukturen dominiert (Abbildung 2). Die hellen Zellen sind pflanzenähnlich und mit glykogen gefüllt. Im Weiteren finden sich Nekrosen und Blutungen.
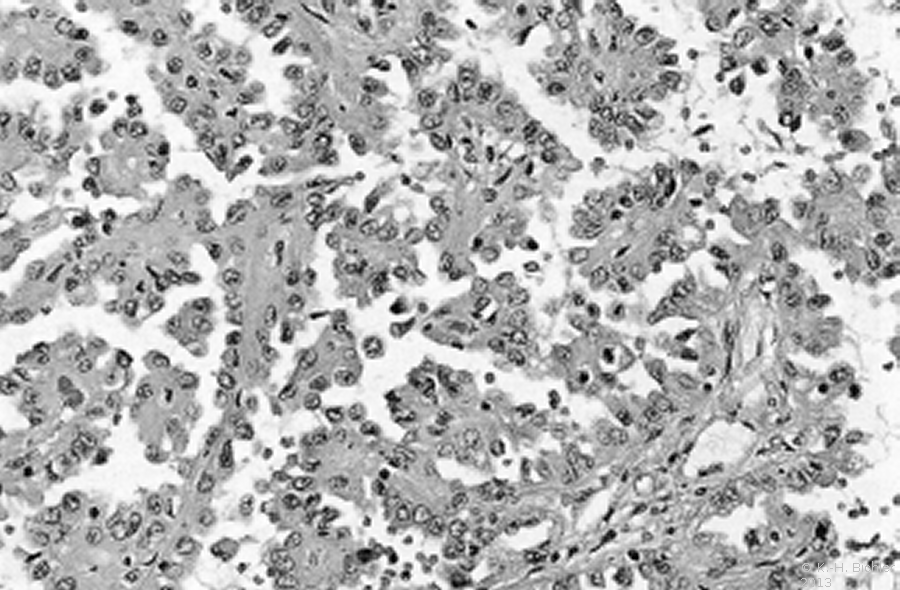
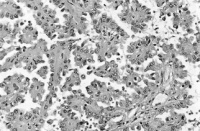
Hierbei sind die Tumorzellen in papillärer Struktur angeordnet (Abbildung 3).
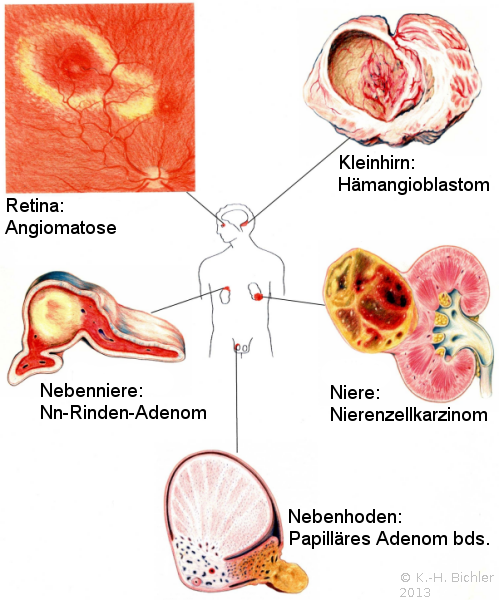
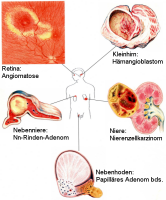
Nierenzellkarzinome im Kindesalter treten als Bestandteil des von Hippel-Lindau-Syndroms, zum Teil auch beidseitig, auf (Abbildung 4) ![]() Literatur:Salome, F. et al: "Renal Lesions in von Hippel-Lindau Disease: The Benign, the Malignent, the Unknown", Eur Urol 34, 383-392, 1998. Das von Hippel-Lindau-Syndrom ist eine autosomal-dominant vererbte Anlagestörung der embryonalen Gefäßorganisation (Angioblastome). Ursache sind Mutationen eines Gens im Chromosom 3. Durch diese Veränderung wird ein Protein nicht mehr gebildet, das eine Schutzfunktion gegenüber Gefäßen, Drüsen und Nierenzellen hat. Knaben sind häufiger betroffen als Mädchen. Die Prävalenz liegt zwischen 1:20.000 und 1:50.000
Literatur:Salome, F. et al: "Renal Lesions in von Hippel-Lindau Disease: The Benign, the Malignent, the Unknown", Eur Urol 34, 383-392, 1998. Das von Hippel-Lindau-Syndrom ist eine autosomal-dominant vererbte Anlagestörung der embryonalen Gefäßorganisation (Angioblastome). Ursache sind Mutationen eines Gens im Chromosom 3. Durch diese Veränderung wird ein Protein nicht mehr gebildet, das eine Schutzfunktion gegenüber Gefäßen, Drüsen und Nierenzellen hat. Knaben sind häufiger betroffen als Mädchen. Die Prävalenz liegt zwischen 1:20.000 und 1:50.000 ![]() Literatur:Neumann, H. P. H.: "Von-Hippel-Lindau-Syndrom unterschätzt und häufig verkannt", DÄB, 90, B571-587, 1993. Von Rahner und Steinke wird die Häufigkeit mit 1:36.000 angegeben. Dabei bezieht sich diese auf die Zahl der Anlageträger in der Allgemeinbevölkerung
Literatur:Neumann, H. P. H.: "Von-Hippel-Lindau-Syndrom unterschätzt und häufig verkannt", DÄB, 90, B571-587, 1993. Von Rahner und Steinke wird die Häufigkeit mit 1:36.000 angegeben. Dabei bezieht sich diese auf die Zahl der Anlageträger in der Allgemeinbevölkerung ![]() Literatur:Rahner, N., Steinke, V.: "Erbliche Krebserkrankungen", DÄB, 105, 706-715, 2008. Die Läsionen an der Retina (Angiomatosus retinae- v. Hippel) und das Hämangioblastom des ZNS -im Kleinhirn meist zystisch- Lindau Tumor bezeichnen das Krankheitsbild: von Hippel-Lindau-Syndrom (Abbildung 4)
Literatur:Rahner, N., Steinke, V.: "Erbliche Krebserkrankungen", DÄB, 105, 706-715, 2008. Die Läsionen an der Retina (Angiomatosus retinae- v. Hippel) und das Hämangioblastom des ZNS -im Kleinhirn meist zystisch- Lindau Tumor bezeichnen das Krankheitsbild: von Hippel-Lindau-Syndrom (Abbildung 4) ![]() Literatur:Ekman, P. Damber, J.-E.: "Hereditary and Familial Malignancies of the Urogential Tract", European Urology Update Series, 7, 124-131, 1998.
Literatur:Ekman, P. Damber, J.-E.: "Hereditary and Familial Malignancies of the Urogential Tract", European Urology Update Series, 7, 124-131, 1998.
Beginnend im 3. Embryonalmonat, in dem die Retina vaskularisiert wird, wird die Erkrankung meist erst im 2. Lebensjahrzehnt manifest. Es finden sich polytope angioblastische Fehlbildungen (Retina, ZNS, Nebenniere und Nebenhoden), häufig kombiniert mit viszeralen zystischen und tumorösen Veränderungen (Nierenzellkarzinome, Pankreaskarzinome, Phäochromozytome). Das Nierenzellkarzinom stellt die häufigste Todesursache dar ![]() Literatur:Maher, E. R., Yates, J. R. W., Harris, R. et al: "Clinical features and natural history of von Hippel-Lindau disease", Q J Med 77: 1151-1163, 1990. Symptomatisch werden die Patienten durch Bluthochdruck (Phäochromozytom!), Gangstörungen bzw. überraschende Erblindung.
Literatur:Maher, E. R., Yates, J. R. W., Harris, R. et al: "Clinical features and natural history of von Hippel-Lindau disease", Q J Med 77: 1151-1163, 1990. Symptomatisch werden die Patienten durch Bluthochdruck (Phäochromozytom!), Gangstörungen bzw. überraschende Erblindung.