Phäochromozytom
- a
b 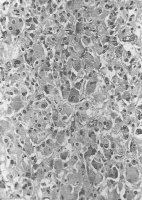
-
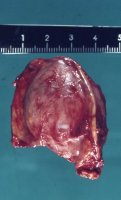
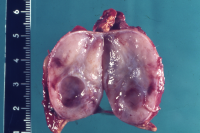
- Abbildung 1:
-
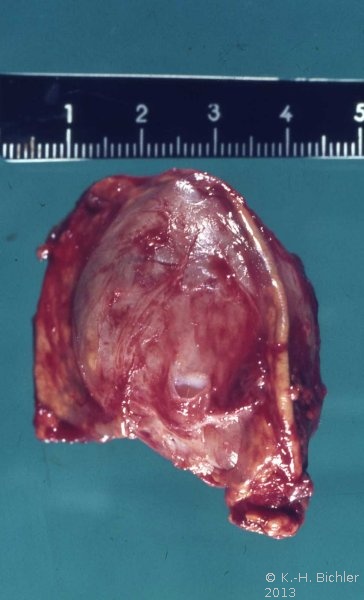
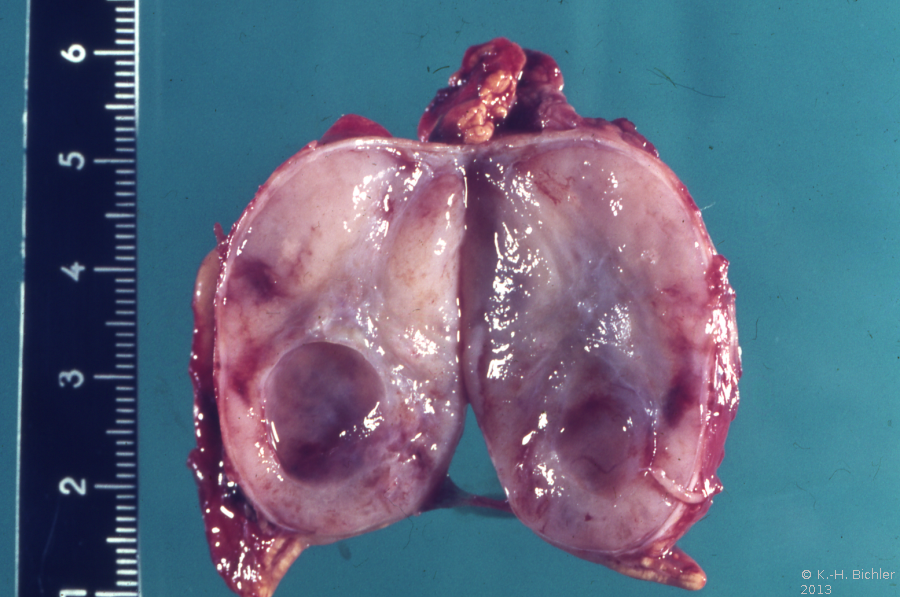
- Nebennierentumor links bei 9 Jahre altem Jungen: 3,5x4,5cm großer gekapselter Tumor von graugelblicher Farbe. Die Schnittfläche zeigt einen zystenartigen Hohlraum. Erhaltenes Nebennierengewebe kaum mehr erkennbar, Reste am oberen und unteren Bildrand
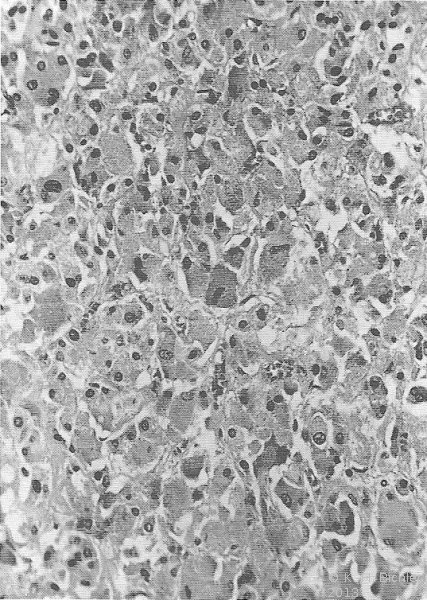
- Histologie: Trabekulierte Struktur mit großen, vielgestaltigen, irregulär angeordneten Zellen bzw. mehrgestaltigen Zellkernen (Pleomorphie). Sowie einzelne Zellgruppen, von Bindegewebestroma umfasst
Der seltene Tumor tritt mit einer Inzidenz von 1 zu 8 Millionen auf. Der Häufigkeitsgipfel liegt im mittleren Lebensalter (40 - 60 Jahre). Bei Kindern wird der Tumor öfters bilateral beobachtet (s. Kasuistik). Jungen sind häufiger betroffen als Mädchen. Die Tumoren treten aber auch familiär (s.u.) gebunden auf.
Ca. 90% sind solitär, etwa 10% in Syndromen wie Hippel-Lindau ( s. Niere/Nierenzellkarzinom/Abbildung 4), Neurofibromatose/von Recklinghausen, Sturge-Weber bzw. Medullary Thyroidcarcinoma (MEN II,III).
s. Niere/Nierenzellkarzinom/Abbildung 4), Neurofibromatose/von Recklinghausen, Sturge-Weber bzw. Medullary Thyroidcarcinoma (MEN II,III).
Allgemein formuliert ist festzustellen, dass der Tumor in 10% bilateral, 10% extraadrenal, 10% maligne und in 10% bei Kindern und Jugendlichen auftritt (von Lack deshalb als 10%-Tumor bezeichnet)  Literatur:Lack, E. E. et al: "Adrenal Glands" in Bostwick, D. G., Eble, J. N.: "Urologic Surgical Pathology", Mosby St. Louis, 1997.
Literatur:Lack, E. E. et al: "Adrenal Glands" in Bostwick, D. G., Eble, J. N.: "Urologic Surgical Pathology", Mosby St. Louis, 1997.
Morphologie
Ca. 85% der Tumoren entstehen im Mark der Nebenniere (Medulla). Es handelt sich um scharf umschriebene, gekapselte, in der Regel gutartige Tumoren unterschiedlicher Größe (1-2 bzw. bis 4-5 cm) (s. Kasuistik). Vereinzelt auch über faustgroß (Nebennierenkarzinom).
Die Tumoren zeigen auf der Schnittfläche z.T. blutigen Zerfall bzw. zystische Aussparungen (Abbildung 1a).
Histologie: Der Tumor ist aus chromaffinen Zellen aufgebaut, die Katecholamine und Peptidhormone sezernieren (Abbildung 1b).
Eine maligne Entartung wird in 15-25% beschrieben 10-15% der Tumoren finden sich extraadrenal in den Ganglien des sympatischen N.S., bei Kindern bis zu 35% Literatur:Mundschenk, J. Dietrich, K. D et al: "Phöochromozytom. Klinik, Diagnostik und Therapie", DÄB 98, 39, A2502-2510, 2001.
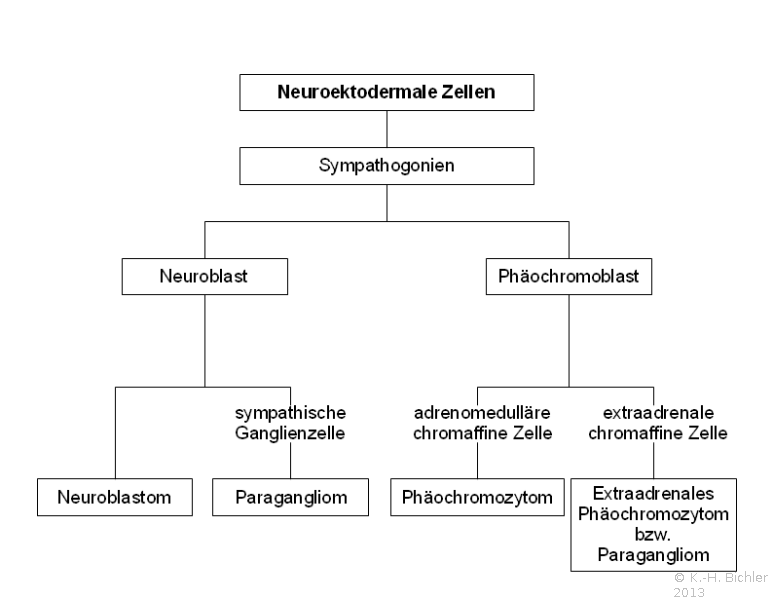
Die Tumoren entwickeln sich aus neuroektodermalen Zellen - Embryogenese ( Abbildung 2).
Abbildung 2).