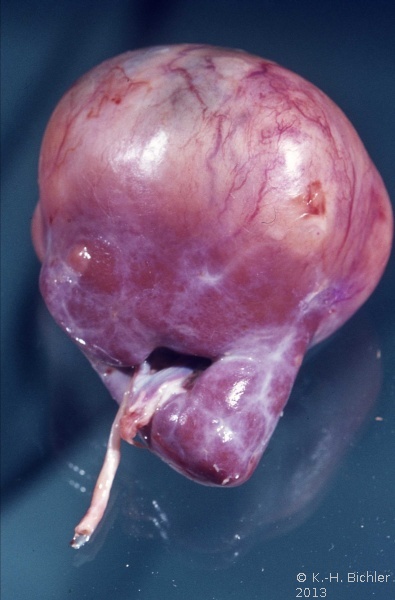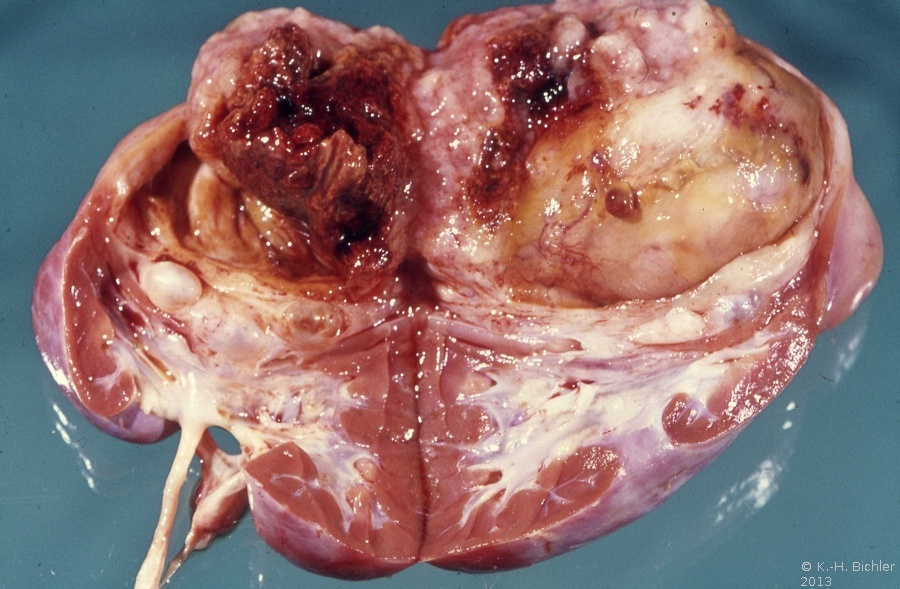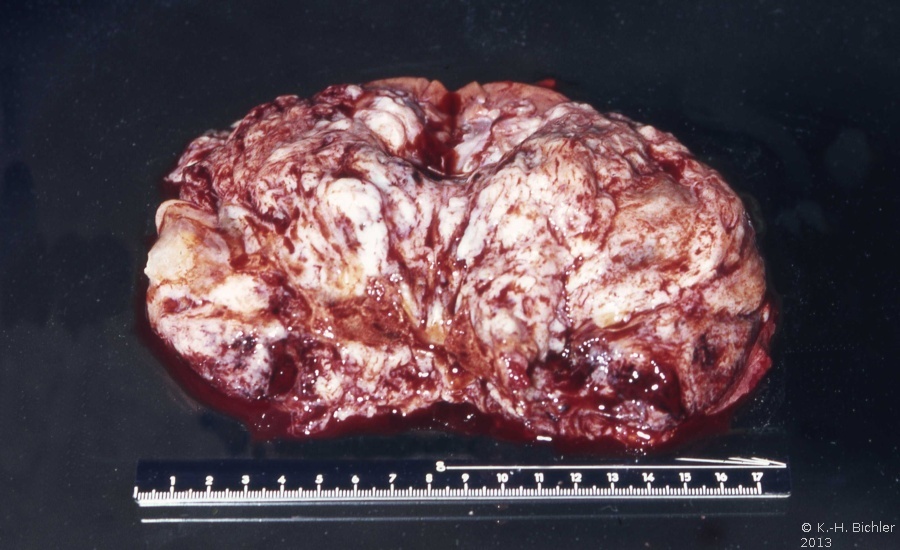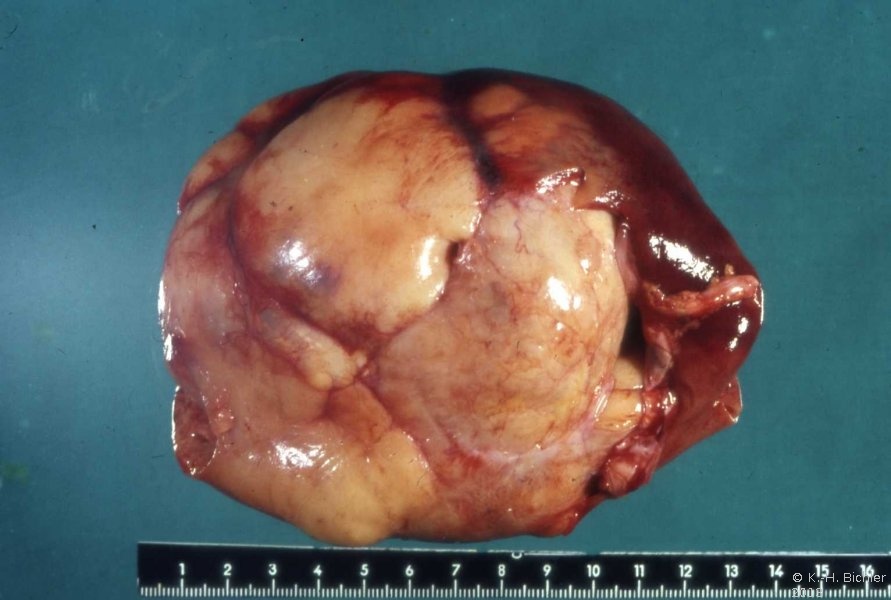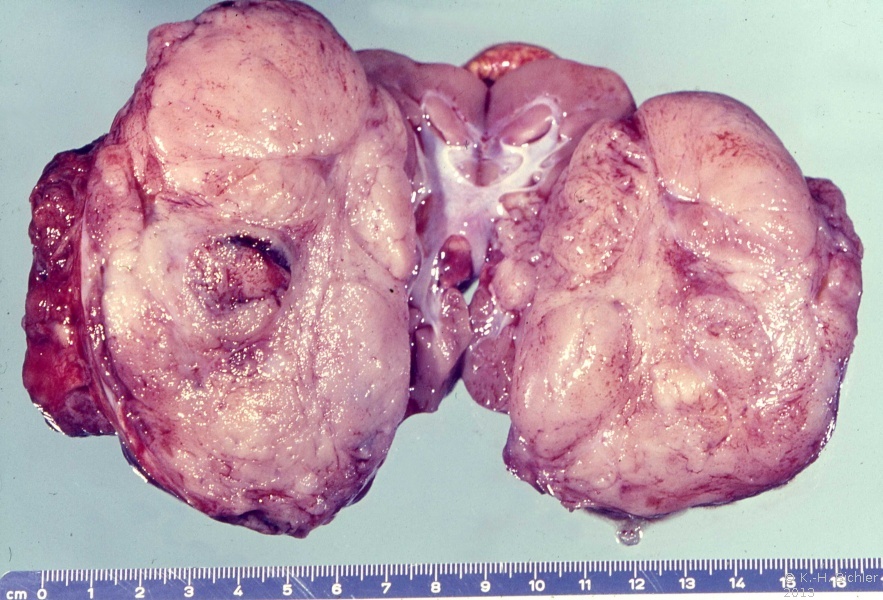
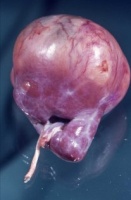
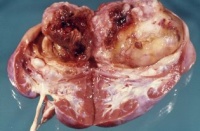
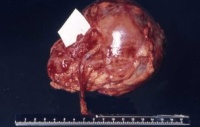
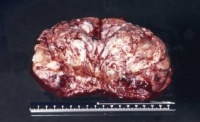
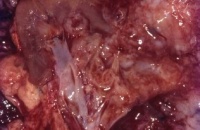
- Abbildung 1: Wilmstumoren von Kindern im Alter von 8 Monaten bis 4 Jahren
- a) Nahezu die ganze Niere einnehmender, grauweißlicher Tumor von glatter Oberfläche, z.T. knollige Form (1,5 Jahre alter Junge)






 c
c 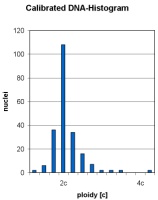
Die karzinomatöse Entartung persistierender metanephrogener Stammzellen (Blastem) auf der Grundlage einer zytogenetischen Deletion auf dem kurzen Arm des Chromosoms 11 in der p13-Region (Wilmstumor1-Gen) bzw. Mutation auf 11p15,5 (WT2-Gen) führen zur Entwicklung eines Wilmstumors. Durch die Pluripotenz des Blastems können vielfältige epitheliale und mesenchymale Zell- und Gewebskomponenten nebeneinander histologisch nachweisbar sein, wie beispielsweise alle in der Niere vorkommenden Gewebe, aber auch Skelettmuskel-, Knorpel- und Schleimzellen. Die Tumoren können zystisch aufgebaut sein.
Die karzinomatöse Entartung persistierender metanephrogener Stammzellen (Blastem) auf der Grundlage einer zytogenetischen Deletion auf dem kurzen Arm des Chromosoms 11 in der p13-Region (Wilmstumor1-Gen) bzw. Mutation auf 11p15,5 (WT2-Gen) führen zur Entwicklung eines Wilmstumors. Durch die Pluripotenz des Blastems können vielfältige epitheliale und mesenchymale Zell- und Gewebskomponenten nebeneinander histologisch nachweisbar sein, wie beispielsweise alle in der Niere vorkommenden Gewebe, aber auch Skelettmuskel-, Knorpel- und Schleimzellen. Die Tumoren können zystisch aufgebaut sein. Anaplastische Tumoranteile (Kernhyperchromasie, hyperdiploide Mitosen, mehr als dreifach vergrößerte Zellkerne) treten in ca. 5% der Fälle auf und sind als prognostisch ungünstiges Kriterium zu werten. Die Tumorzellen können - genau wie beim Nierenzellkarzinom - durch Aggregation in den Blutgefäßen Tumorthromben bilden (Abbildung 1c). Wilmstumoren metastasieren nach medial in die Lymphknoten und hämatogen in die Lunge. Vereinzelt finden sich ZNS-, Leber- und Skelettmetastasen.
Der Tumor verdrängt zumeist deutlich abgegrenzt das residuale Nierengewebe.
Auf der Schnittfläche ist der Tumor von gelbbrauner bis grauer Farbe mit einzelnen Einblutungen. Zystische Anteile und Nekrosen kommen vor sowie oberflächlich glatte, knotenförmige Strukturen, ähnlich einem Rhabdomyosarkom (Abbildung 1a-d).
(Prof. Dr. E. Schäfer, Freiburg)
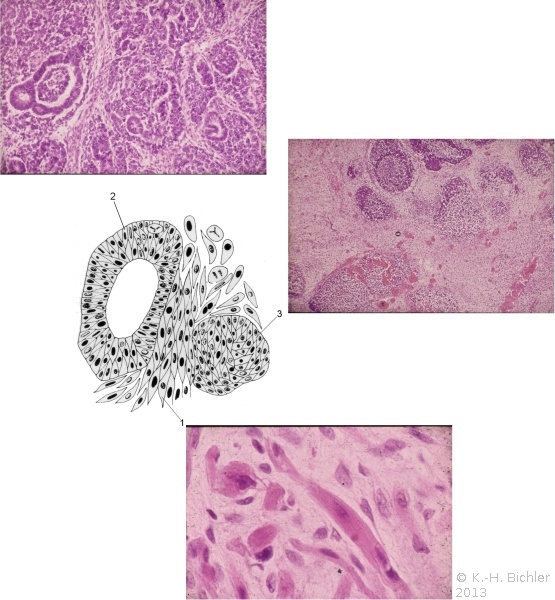
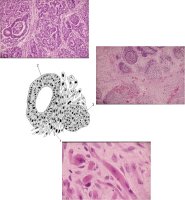
Mikroskopisch ist der Wilmstumor gekennzeichnet durch Gewebe, das verschiedenen Stufen der Nephrogenese entspricht. Es finden sich blastemische, stromale und epitheliale Zelltypen. Zum epithelialen Anteil gehören Strukturen, die tubulär oder glomerulär aufgebaut sind. Fibrozyten und myxoide Formen gehören zu den stromalen Gewebsarten. Auch glatte Muskelzellen, Knorpel-, Fett- und neurales Gewebe finden sich. Grundsätzlich ist festzuhalten, dass der Wilmstumor histologisch heterogen bzw. triphasisch aufgebaut ist. Die Anteile der Komponenten sind verschieden und korrelieren mit der Malignität des Tumors (Abbildung 2).
Tumoren des Stadium I (begrenzt auf die Niere) sind zumeist aus epithelialen Strukturen aufgebaut. Die vornehmlich aus blastomatösem Gewebe bestehenden Geschwülste gehören zu den Stadien III und IV (Stadieneinteilung s. später). Ungefähr 5% der Wilmstumoren enthalten anaplastische Nester. Die Zellen enthalten hyperchrome, große, pleomorphe (polymorphe) Kerne mit abnormalen Mitosen. Diese Tumoren sprechen im allgemeinen schlecht auf die Chemotherapie an und sind nicht komplett resezierbar (Abbildung 2).
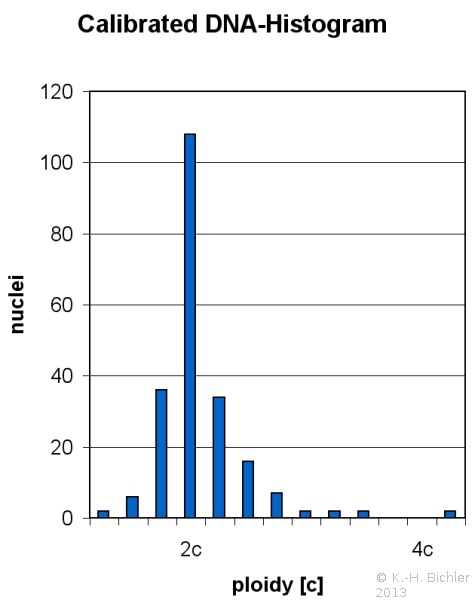
Die Beziehungen zwischen dem Wilmstumor und anderen nephroblastomatösen Tumoren wie dem zystisch partiell differenzierten Nephroblastom, dem zystischen Nephrom, der Nephroblastomatosis und dem multilokulären, zystischen Nierenzellkarzinom sind nicht eindeutig und bleiben kontrovers (Abbildung 3).
Eine in Beziehung zum Wilmstumor stehende Geschwulstform ist das Zystische Nephrom.
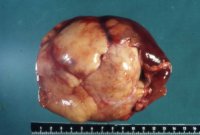
Es handelt sich um einen seltenen benignen Tumor. Er tritt bei Jugendlichen bzw. im frühen aber auch höheren Erwachsenenalter häufiger bei Frauen auf. Bei Kindern unter zwei Jahren öfter bei männlichen. Es werden aber auch Tumoren im höheren Erwachsenenalter festgestellt. Makroskopisch ist der Tumor gut umschrieben und durch eine fibröse Kapsel vom normalen Nierengewebe abgetrennt.
Auf der Schnittfläche stellen sich zystenartige Veränderungen und solide Areale dar (Abbildung 4a). Histologisch erkennt man multiple Zysten und Septen aus fibrösem Gewebe. Die Zysten sind von einem flachen, kubischen Epithel ausgekleidet (Abbildung 4b). Die Zytometrie zeigt einen diploiden DNA-Gehalt (Abbildung 4c).
Im Zusammenhang mit der Heterogenität bzw. der molekularen Genetik der Wilmstumoren ist das Vorkommen einer prämalignen Struktur (Präkanzerose), der "Nephroblastomatosis" zu nennen. Es handelt sich um multizentrische, bzw. diffuse Nester von unreifem, nephrogenem Gewebe in Bezirken von normalen Nierenanteilen. Die Nephroblastomatosis kann in ein malignes Nephroblastom übergehen (s. Abbildung 3).
Zwei Tumoren der Niere, die ebenfalls im Kindesalter auftreten, sind das eher gutartige, kongenitale mesoblastische Nephrom und der hochmaligne Rhabdoid-Tumor.
Das mesoblastische Nephrom wird im Säuglingsalter im 3.-6. Monat entdeckt, zumeist durch einen Oberbauchtumor ![]() Literatur:Richmond, H. et al: "Neonatal Renal Tumors", J Pediatr Surg, 5, 413-417, 1970. Der Tumor ist oberflächlich glatt, z.T. bucklig, von weißgrauer Farbe (s. Abbildung 1d). Ein von der Makroskopie her vergleichbares Bild wie bei dem Wilmstumor des 8 Monate alten Mädchens. Histologisch unterscheidet sich das Nephrom vom Wilmstumor, der zumeist ältere Kinder betrifft. Es finden sich dicht verflochtene Bündel von Spindelzellen mit elongierten Kernen, die das renale und perineale Gewebe infiltrieren, Einschlüsse von Glomeruli und Tubuli in das Tumorgewebe sind häufig. In wie weit das mesoblastische Nephrom zur Gruppe der Wilmstumoren gehört, ist fraglich. Die Prognose des mesoblastischen Nephroms ist gut. Metastasen sind selten
Literatur:Richmond, H. et al: "Neonatal Renal Tumors", J Pediatr Surg, 5, 413-417, 1970. Der Tumor ist oberflächlich glatt, z.T. bucklig, von weißgrauer Farbe (s. Abbildung 1d). Ein von der Makroskopie her vergleichbares Bild wie bei dem Wilmstumor des 8 Monate alten Mädchens. Histologisch unterscheidet sich das Nephrom vom Wilmstumor, der zumeist ältere Kinder betrifft. Es finden sich dicht verflochtene Bündel von Spindelzellen mit elongierten Kernen, die das renale und perineale Gewebe infiltrieren, Einschlüsse von Glomeruli und Tubuli in das Tumorgewebe sind häufig. In wie weit das mesoblastische Nephrom zur Gruppe der Wilmstumoren gehört, ist fraglich. Die Prognose des mesoblastischen Nephroms ist gut. Metastasen sind selten ![]() Literatur:Richmond, H. et al: "Neonatal Renal Tumors", J Pediatr Surg, 5, 413-417, 1970
Literatur:Richmond, H. et al: "Neonatal Renal Tumors", J Pediatr Surg, 5, 413-417, 1970
Retik, A.B.: "Congenital mesoblastic nephroma" in Seidman, E.J. (ed.): "Current Urologic Therapy", Saunders Philadelphia, 1994
Eble, J.N.: "Neoplasms of the kidney" in Bostwick, D.G, Eble, J.N: "Urologic Surgical Pathology", Mosby St. Louis, 1997. Die Behandlung besteht in der Nephrektomie. Chemotherapie ist aufgrund der günstigen Prognose verzichtbar, wobei mit Retik festzuhalten ist, dass die Prognose für Wilmstumore, die im ersten Lebensjahr festgestellt werden, ebenfalls günstig ist. Hier erhebt sich die Frage in wie weit ein Teil davon als nephroblastisches Nephrom anzusehen ist ![]() Literatur:Retik, A.B.: "Congenital mesoblastic nephroma" in Seidman, E.J. (ed.): "Current Urologic Therapy", Saunders Philadelphia, 1994.
Literatur:Retik, A.B.: "Congenital mesoblastic nephroma" in Seidman, E.J. (ed.): "Current Urologic Therapy", Saunders Philadelphia, 1994.
Zu den malignen Neubildungen der Niere gehört fernerhin der bei weitem bösartigste ![]() Rhabdoid-TumorRhabdoid-TumorICD-0 8963/3. Dieses Malignom metastasiert frühzeitig und führt häufig binnen Jahresfrist zum Tode. Die Tumoren sind nicht gekapselt und infiltrieren das Nierenparenchym und -becken. Histologisch sind sie aus polygonalen Zellen mit prominenten Nucleoli aufgebaut. Auffällig sind zytoplasmatische Einschlüsse, die die Kerne verdrängen
Rhabdoid-TumorRhabdoid-TumorICD-0 8963/3. Dieses Malignom metastasiert frühzeitig und führt häufig binnen Jahresfrist zum Tode. Die Tumoren sind nicht gekapselt und infiltrieren das Nierenparenchym und -becken. Histologisch sind sie aus polygonalen Zellen mit prominenten Nucleoli aufgebaut. Auffällig sind zytoplasmatische Einschlüsse, die die Kerne verdrängen ![]() Literatur:Eble, J.N.: "Neoplasms of the kidney" in Bostwick, D.G, Eble, J.N: "Urologic Pathologic Surgery", Mosby St. Louis, 1997.
Literatur:Eble, J.N.: "Neoplasms of the kidney" in Bostwick, D.G, Eble, J.N: "Urologic Pathologic Surgery", Mosby St. Louis, 1997.