- Abbildung 2:
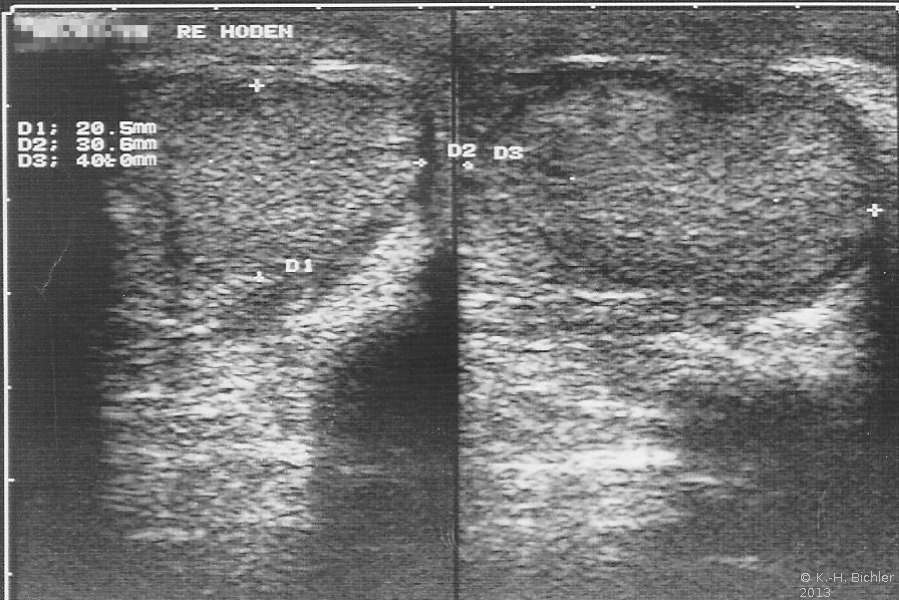
- In der Sonographie rechter Hoden von normaler Größe


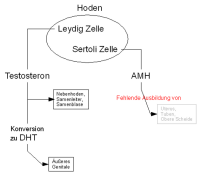
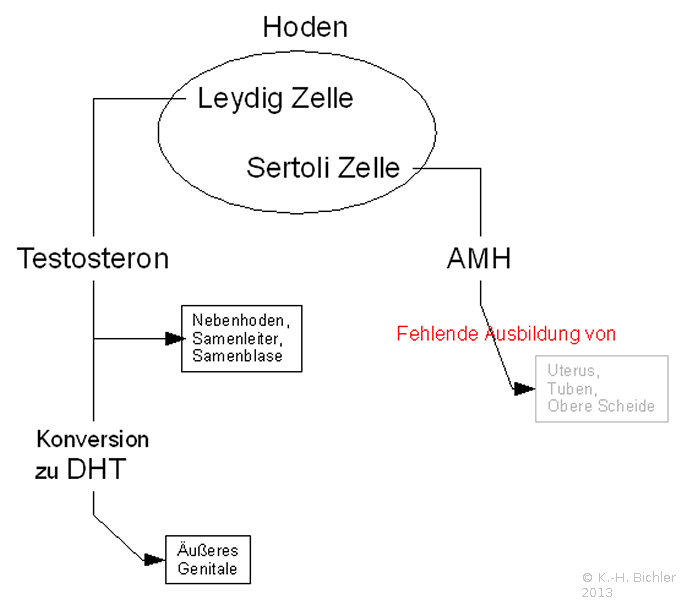
Die normale männliche Entwicklung erfordert eine adäquate Differenzierung der Hoden in der Fetalperiode, Synthese und Ausschüttung der testikulären Hormone und entsprechende Antwort der Zielorgane auf diese Hormone. Das Anti-Müller-Gang-Hormon wird von den Sertoli-Zellen gebildet und inhibiert die Entwicklung der Müller-Gang-Derivate (Uterus, Tuben und Teile der Scheide). Das von den Leydig-Zellen produzierte Testosteron bewirkt die Differenzierung der Wolff-Gänge in die männlichen Genitalgänge. Die Konversion von Testosteron zu Dehydrotestosteron durch das Enzym 5-α-Reduktase sichert die Entwicklung des männlichen äußeren Genitale (Abbildung 1). Störungen in diesen Prozessen können zum männlichen Pseudohermaphroditismus führen ![]() Literatur:Nistal, M. et al: "Non-Neoplastic diseases of the testis" in Bostwick, D. G. et al: "Urologic Surgical Pathology", Mosby St. Louis, 1997
Literatur:Nistal, M. et al: "Non-Neoplastic diseases of the testis" in Bostwick, D. G. et al: "Urologic Surgical Pathology", Mosby St. Louis, 1997
Neher, R. et al: "Steroid transformations in vitro by testicular tissue from two cases of testicular feminisation", Acta Endocrin., 29, 177-192, 1965.
Die Geschlechtsdetermination benötigt ein Virilisierungsgen SRy. Die Virilisierung des fetalen Genitaltraktes hängt von testikulären Hormonen ab: Testosteron und Anti-Müller-Gang-Hormon (AMH). Bei einer testikulären Dysgenesie oder einem echten Hermaphroditismus (heterogene gonadale Differenzierung) fehlen Testosteron und AMH. Persistierende Müller-Gänge und Hypospadie sind in diesem Zusammenhang klinisch am häufigsten. Das zeitliche Einsetzen der testikulären Degeneration beeinflusst das klinische Bild. So bilden sich die Müller-Gänge zurück, wenn der Hoden bis zu 8 Wochen in normaler Funktion bleibt.
Störungen der testikulären Differenzierung werden nur durch ein Hormon beeinflusst.
Isolierte Defekte von AMH oder seinem Rezeptor sind relativ selten. Sie können durch Bestimmung von AMH, das niedrig oder nicht messbar ist, erfasst werden. Das Fehlen von AMH ist zumeist Folge einer Mutation des AMH-Gens.
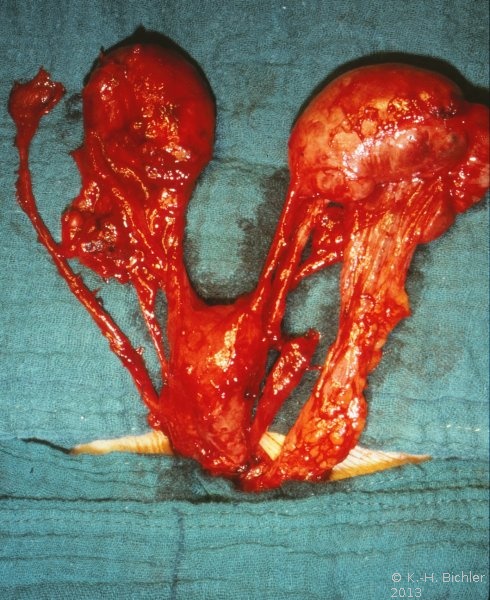
Ein Beispiel männlichen Pseudohermaphroditismus' aus der Gruppe von Krankheitsbildern infolge einer gestörten Rückbildung der Müller-Gänge wegen Fehlens von AMH ist das Syndrom des persisterienden Müller-Ganges (PMDS). Andere Bezeichnungen dafür sind "Male with Uterus", "Tubular Hermaphroditism", "Persistend Oviduct Syndrome" und "Hernia uteri inguinalis". Dabei handelt es sich um die charakteristische Form der isolierten AMH-Defizienz. PMDS ist eine seltene Form des männlichen Pseudohermaphroditismus mit Derivaten der Müller-Gänge bei einem phänotypisch normalen Mann ![]() Literatur:Delaney, D. P. et al: "Persistent Mullerian Duct Snydrome Associated with 47,XXY Genotype", J Urol, 171, 852-853, 2004.
Literatur:Delaney, D. P. et al: "Persistent Mullerian Duct Snydrome Associated with 47,XXY Genotype", J Urol, 171, 852-853, 2004.
Die äußeren Genitalien sind männlich, allerdings sind in 25% ein Hoden und in 75% beide kryptorch. Häufig findet sich eine inguinale Hernie, deren Inhalt aus Uterus, Tuben und Teilen der Vagina besteht. Gewöhnlich liegt Infertilität vor. Die Hoden neigen zur malignen Entartung.
Bei einzelnen Patienten mit diesem Syndrom sind die kryptorchen Hoden in ektoper Lage mit den Derivaten der Müller-Gänge verwachsen (Kasuistik).
 b
b 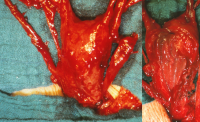
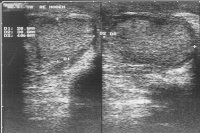
Das Beispiel zeigt die Situation bei einem 25 Jahre alten Mann, der phänotypisch normal männlich war. Bei der Untersuchung war der rechte Hoden tastbar und sonographisch nachweisbar (Abbildung 2).
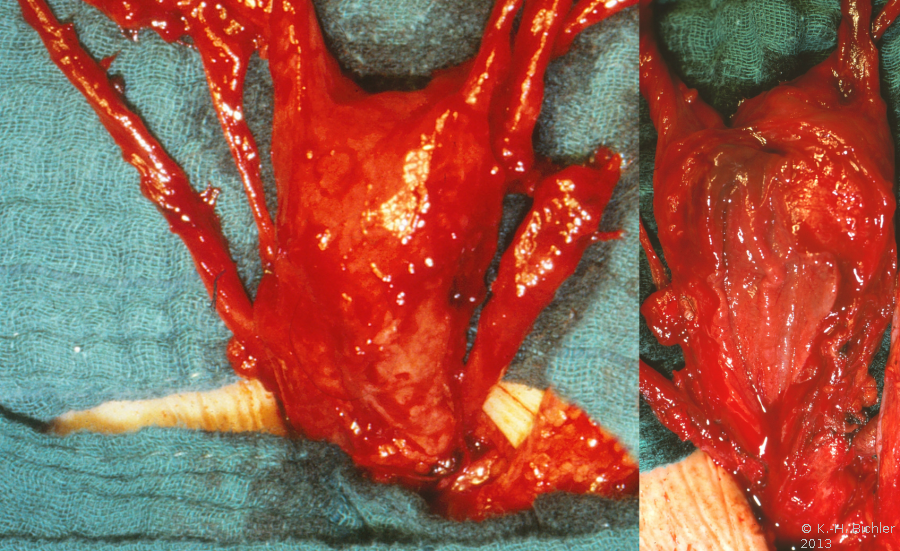
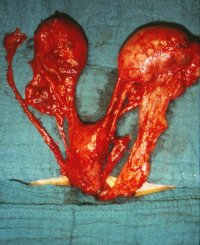
Links kein Hoden palpabel. Verdacht auf Hodendeszensusstörung. Wegen einer rechtsseitigen Hernie wurde die Leiste von einem Inguinalschnitt aus freigelegt. Dabei fanden sich zwei Hoden verbacken mit einem rudimentären Uterus, tubulären Strukturen und einem Vaginalstrang (Abbildung 3). Der kleinere Hoden sowie die Müller-Gang-Derivate wurden entfernt.
Testosteron- bzw. Dihydrotestosteronmangel oder Fehlen der Hormone ist Folge gestörter Leydig-Zell-Aktivität (Impaired Leydic Cell Activity), Androgensynthesestörung, bzw. 5-α-Reduktase-Mangel.
Eine gestörte Testosteronsynthese kann durch Abnormitäten von Enzymen verursacht werden, die für die Pregnenolonbildung notwendig sind: 3-β-hydroxysteroid dehydrogenase; 17-α-hydroxylase; 17,20 desmolase; and 17-β-hydroxysteroid dehydrogenase.
5-α-Reduktase-Mangel findet sich häufiger in türkischen Populationen infolge homozysgoter Missenzmutation des 5-α-Reduktase-Typ-2-Gen ![]() Literatur:Bahceci, M. et al: "A novel missens mutation of 5-α-reduktase type 2 gene (SRD5A2) leads to severe male pseudohermaphroditism in a turkish family", Urology 66: 407-410, 2005.
Literatur:Bahceci, M. et al: "A novel missens mutation of 5-α-reduktase type 2 gene (SRD5A2) leads to severe male pseudohermaphroditism in a turkish family", Urology 66: 407-410, 2005.
Diese autosomal rezessiven Syndrome sind durch Fehler in der Testosteronbiosynthese charakterisiert und gekennzeichnet durch unvollständige oder fehlende Virilisation.
Infolge gestörter Testosteronbiosnythese bzw. 5-α-Reduktase-Mangel treten regulär weiblich ausgeprägte äußere Geschlechtsteile auf, aber auch regulär ausgebildete männliches Genitale mit Uterus, Tuben und Teilen der Scheide in einer hernia Inguinalis kommen vor (s. Kasuistik).
Das klinische Bild (Phänotyp) ist unterschiedlich und zwar von nur geringgradigen Abweichungen des männlichen Genitale bis zum vollständigen weiblichen Phänotyp ![]() Literatur:Hiort, O. et al:"Androgenresistenzsyndrome - Klinische und molekulare Grundlagen", DÄB 96, B-526-B531, 1999.
Literatur:Hiort, O. et al:"Androgenresistenzsyndrome - Klinische und molekulare Grundlagen", DÄB 96, B-526-B531, 1999.
Rekonstruktive Maßnahmen bei männlichem Pseudohermaphroditismus sind bei Fehlbildungen des äußeren männlichen Genitaltraktes wie Mikropenis, Kryptorchismus (s. Kasuistik) bzw. Hypospadie angezeigt.
Hiort et al empfehlen in Hinsicht auf das klinische Vorgehen in Anlehnung an die Einteilung von Prader beim adrenogenitalen Syndrom 5 verschiedene Grade (![]() s. Einleitung/Abbildung 3).
s. Einleitung/Abbildung 3).
Im Weiteren sind hier Krankheitsbilder zu erwähnen, denen eine mehr oder weniger ausgebildete Androgenresistenz (Androgene insensivity syndrome - AIS) zugrunde liegt. Dabei handelt es sich um eine ungenügende Bindung von Androgenen an nukleäre bzw. zelluläre Rezeptoren.
Verschiedene klinische Formen sind zu unterscheiden, die vom normalen männlichen Phänotyp (Penis und Skrotum mit Hoden) bis vollständig normalen weiblichen äußeren Genitale reichen.
Auffällig sind bei einigen dieser Kinder mit normalem weiblichen äußeren Genitale die Ausbildung ein- oder beiderseitiger inguinaler Hernien, die männliche Gonaden enthalten (Complete androgen insensitivity - CAIS). Als Leitsymptom: Inguinale Hernie bei Mädchen (Kariotyp XY!) ![]() Literatur:Deeb, A., Huhges, I. A.: "Inguinal Hernia in female infants: a cue to check the sex chromosomes?", BJU int, 96, 401-403, 2005.
Literatur:Deeb, A., Huhges, I. A.: "Inguinal Hernia in female infants: a cue to check the sex chromosomes?", BJU int, 96, 401-403, 2005.