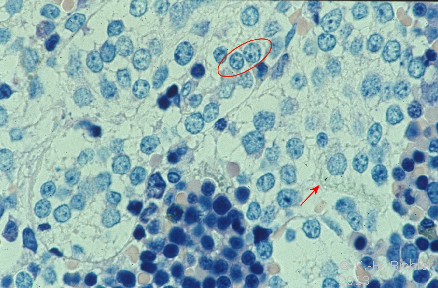
- Abbildung 1:
- Neuroblastomzellen. Markiert Kerne mit zerstreutem Chromatin und Zytoplasma mit Neurofilamenten mit unscharfer Begrenzung
- (Die Abbildung verdanken wir Prof. Dr. H. E. Schaefer, Freiburg)


Im Kindesalter gehen 25 bis 40% der Neuroblastome vom Nebennierenmark aus, andere treten im sympathischen Nervensystem, insbesondere der paravertebralen Region des hinteren Mediastinum bzw. des unteren Abdomens und des Beckens auf (in allen Regionen des Grenzstrangs).
Der Tumor kann als kleiner Knoten (in situ) auftreten oder erhebliche Ausmaße annehmen (1Kg!). Die kleinen, verborgenen Formen sind häufiger. Der größte Teil davon unterliegt späterer Regression. Zurück bleibt beim Erwachsenen ein fibrotischer bzw. kalzifizierter Herd. Auch der Übergang (Differenzierung in ein Ganglioneurom) ist möglich. Neben diesen abgekapselten Formen (Neuroblastoma in situ) finden sich infiltrativ wachsende und dabei in die Nachbarschaft, z.B. die Nieren, Nierenvene und/oder Vena Cava eindringende Tumoren (s. Abbildung 2b). Die Neuroblastome sind auf der Schnittfläche gekennzeichnet durch ein graues, gehirnartiges Gewebe mit Einblutungen, Nekrosen und zystischen Anteilen.
Die Neuroblastomzellen zeigen Zellkerne mit zerstreutem Chromatin ("Salt and pepper") sowie strähnig angeordnete Neurofilamente des Zytoplasma (Abbildung 1).